Advancements in drug discovery are revolutionizing the way chemists create new medicines. Researchers have developed a machine learning-based system that enhances the efficiency and cost-effectiveness of creating new molecules. This innovative approach promises to shift the paradigm in drug development.
<p>Newswise — Drug discovery operates somewhat like molecular Tetris. Chemists carefully assemble atoms, adjusting and rearranging until they form a molecule that could potentially serve as a new medicine. Typically, improving these molecular structures takes significant time and financial investment.</p>
<p>In a recent study, researchers leveraged machine learning to create a <a href="https://www.nature.com/articles/s41586-026-10239-7">more intelligent prediction system</a> that could accelerate this process while drastically reducing costs.</p>
<p>“We sometimes rely on advanced, physics-based computational chemistry tools to explore novel reactions. However, these tools are often too costly for predicting the thousands of potential new molecules,” explained Simone Gallarati, co-lead author and joint postdoctoral researcher at the University of Utah and the University of California, Los Angeles. “Our goal was to train statistical models that are smart enough to make accurate predictions on untested reactions while minimizing expenses.”</p>
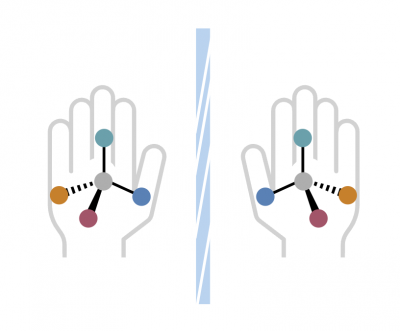
<p>Molecules can exist in two mirror-image forms, a characteristic known as "handedness." The distinction between left-handed and right-handed versions is critical; one variant may provide healing, while the other could be harmful. Chemists must select the appropriate tools—catalysts, ligands, and substrates—to construct the desired version accurately.</p>
<p>The new system functions as a sophisticated filter, capable of screening tens of thousands of chemical structures to predict which versions of a molecule will dominate. This process transforms the components of a reaction into numerical data for computer analysis, thereby establishing the foundation for machine learning predictions.</p>
<p>With minimal input, the model consistently predicted how components would interact, significantly reducing the time, energy, and costs normally associated with laboratory testing.</p>
<p>“Most AI models require vast amounts of data for training, which is a challenge in chemistry since obtaining high-quality, large datasets through experimental work is both expensive and time-consuming,” remarked <a href="https://www.chemistry.utah.edu/faculty/matthew-s-sigman/">Matthew Sigman</a>, a chemist at the University of Utah and co-author of the study. “What’s remarkable about this tool is its ability to leverage smaller data sets, enabling the creation of reasonably accurate models for known reactions and allowing predictions for unfamiliar reactions.”</p>
<p>The study was published as an accelerated preview in the <a href="https://www.nature.com/articles/s41586-026-10239-7">journal Nature</a> on February 11, 2026.</p>
<p><strong>High-Tech Filter</strong></p>
<p>The research team focused their workflow on asymmetric cross-coupling reactions, a powerful strategy for drug development. These reactions connect two carbon-based molecular fragments using a metal catalyst to create more complex compounds. They are termed asymmetric because they favor one handed version of the molecule over the other. Without guidance, experiments tend to yield a 50/50 split. In contrast, asymmetric reactions can produce, for example, 95% of the desired form and only 5% of the unintended mirror image.</p>
<p>Asymmetric cross-coupling reactions typically require three main components: a metal, a ligand, and substrates. The metal catalyst plays a crucial role in binding carbon-based molecules to generate the final product. A ligand attaches to the metal and determines which side of the molecule will react, influencing the three-dimensional orientation of the resultant product. It is arguably the most critical element for controlling a molecule's handedness.</p>
<p>To train their model, Gallarati and their team examined four academic papers on asymmetric reactions, including prior works by coauthors Abigail Doyle and Sigman, that utilized nickel-based catalysts with various ligands. These results served as the sole training data for their workflow. The system was then tasked with predicting outcomes for hypothetical components not included in the initial training data. By progressively challenging the model with increasingly dissimilar materials, the team assessed its predictive capabilities in Doyle's lab, led by Erin Bucci, co-lead author and doctoral student at UCLA.</p>
<p>“As a lab-based chemist, this tool is immensely valuable for saving time on experiments,” Bucci noted. “Instead of conducting 50-60 reactions, we can now run just 5-10, potentially saving weeks or even months. Each component we test requires either purchase or fabrication, so this tool significantly reduces the expense typically associated with acquiring materials.”</p>
<p>Although the authors initially tested the tool for nickel-based reactions, the workflow has far-reaching applications and may deepen our understanding of chemical processes.</p>
<p>“One of the advantages of this workflow is its transparency—it’s not just a black box,” stated <a href="https://doyle.chem.ucla.edu/abby/">Abigail Doyle</a>, a chemist at UCLA and study co-author. “Our predictions offer insights into chemical behavior, even if they are not entirely accurate. We leverage our chemistry knowledge to uncover information we might have missed without this tool.”</p>
<p>The pharmaceutical industry stands to gain significantly from such a tool, as Sigman indicated. For example, if a company needs to produce large quantities of a compound for a clinical trial and wants to apply an established reaction, but it has never been attempted on their specific compound, this tool could prove invaluable.</p>
<p>“The potential for this tool is enormous,” he explained. “Optimizing reactions and managing time and costs is crucial in drug development. This streamlined process could facilitate moving a molecule from phase one to phase two more efficiently.”</p>
<p class="text-center">****</p>
<p>The study has been published in the journal Nature under the title, “Transferable enantioselectivity models from sparse data.” You can access it at <a href="https://www.nature.com/articles/s41586-026-10239-7">https://doi.org/10.1038/s41586-026-10239-7</a>.</p>
<p>The research received support from the Swiss National Science Foundation (#222115), the U.S. National Science Foundation (CHE-2202693 and CHE-1048804), the National Institutes of Health (S10OD028644), and the Center for High Performance Computing at the University of Utah.</p>