Drug discovery resembles a game of molecular Tetris, where chemists piece together atoms, adjusting configurations until a new molecule emerges as a potential medicine. Traditionally, enhancing molecular structures requires significant investment in both time and resources.
However, researchers have taken a significant step forward by utilizing machine learning to develop a more efficient prediction system, dramatically accelerating the drug discovery process at a reduced cost.
“We often use advanced, physics-based computational chemistry tools to analyze novel reactions. Yet, these tools are prohibitively expensive for predicting thousands of potential new molecules,” explained Simone Gallarati, the study’s co-lead author and a joint postdoctoral researcher at the University of Utah and the University of California, Los Angeles. “Our aim was to develop statistical models capable of making precise predictions concerning untested reactions while keeping expenses as low as possible.”
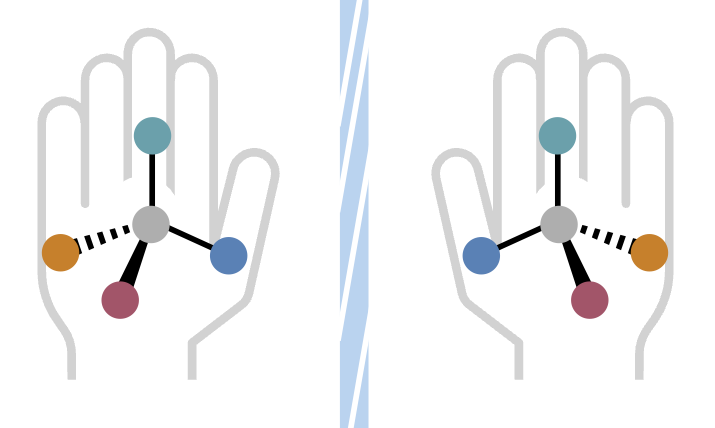
Molecules can exist as mirror images, a phenomenon referred to as “handedness.” The distinction between left and right-handed forms is vital; one variant may offer healing properties, while the other could be harmful. Chemists must utilize the right tools—catalysts, ligands, and substrates—to ensure they synthesize the correct version.
The newly developed system serves as a sophisticated filter, capable of screening thousands of chemical structures to predict how different components will combine, favoring one version of a molecule over another. This workflow provides a cost-effective method to convert reaction components into numerical data for analytical evaluation, laying the groundwork for machine-learning predictions.
With minimal input, the model accurately predicted how the components would interact, significantly reducing the time, energy, and costs associated with laboratory testing.
“Most AI systems require vast amounts of data to train their models. In chemistry, acquiring large, high-quality datasets through experimental work can be very expensive and time-intensive,” noted Matthew Sigman, a chemist at the University of Utah and coauthor of the study. “What’s remarkable about this tool is that it enables researchers to gather smaller datasets, build reasonably accurate models, and make reliable predictions for known reactions, as well as transfer these predictions to new, untested reactions.”
The findings of the study were published as an accelerated preview in the journal Nature on February 11, 2026.
High-tech Filter

The researchers focused their methodology on asymmetric cross-coupling reactions, a crucial technique in drug development. These reactions merge two carbon-based molecular fragments using a metal catalyst to create more intricate compounds. Asymmetric reactions are particularly valuable as they are designed to favor the production of one “handed” version of a molecule. While conventional methods often yield a 50/50 split of both forms, asymmetric reactions can deliver, for example, 95% of the desired variant and only 5% of the less favorable mirror image.
To facilitate their model training, Gallarati and the team selected four academic papers focusing on asymmetric reactions—prior works by coauthors Abigail Doyle and Sigman included. These papers utilized nickel-based catalysts with various ligands, providing the sole training dataset for the workflow. Subsequently, the team challenged the system to predict the outcomes of hypothetical components that were not part of the training data. They gradually introduced more complex tasks that pushed the algorithm to make predictions with materials increasingly different from the original training data. The predictions were ultimately tested in Doyle’s laboratory, under the guidance of Erin Bucci, another co-lead author and doctoral student at UCLA.

“As a lab chemist, this tool is invaluable for minimizing the time spent on experiments,” Bucci remarked. “For instance, instead of conducting 50 to 60 reactions, we can now perform only 5 to 10, potentially saving several weeks or even months. Each component tested in the lab must be either bought or synthesized, and this tool greatly reduces the budget typically required for materials.”
Although the authors evaluated the instrument specifically within the context of nickel-based reactions, the workflow holds potential applications across different fields and could enhance our overall understanding of chemistry.
“A remarkable aspect of this workflow is that it is not a black box,” stated Abigail Doyle, a chemist at UCLA and coauthor of the research. “We can derive insights about the chemistry from the predictions, even if they aren’t accurate. Our chemistry expertise aids in gaining knowledge we would not acquire without this tool.”
This innovative tool could bring immediate advantages to the pharmaceutical industry, Sigman added. For instance, if a company needs substantial quantities of a compound for a clinical trial and aims to utilize a reaction documented in the literature, but it’s untested on their specific compound target.
“This is precisely where this tool can be immensely beneficial,” he emphasized. “Streamlining the reaction optimization and time-cost can be crucial in drug development. This expedited process could make a significant difference in transitioning a molecule from phase one to phase two.”
****
Gallarati, S. et al., Transferable enantioselectivity models from sparse data. Nature (2026). https://doi.org/10.1038/s41586-026-10239-7
This research was supported by the Swiss National Science Foundation (#222115), the U.S. National Science Foundation (CHE-2202693 and CHE-1048804), the National Institutes of Health (S10OD028644), and the Center for High Performance Computing at the University of Utah.